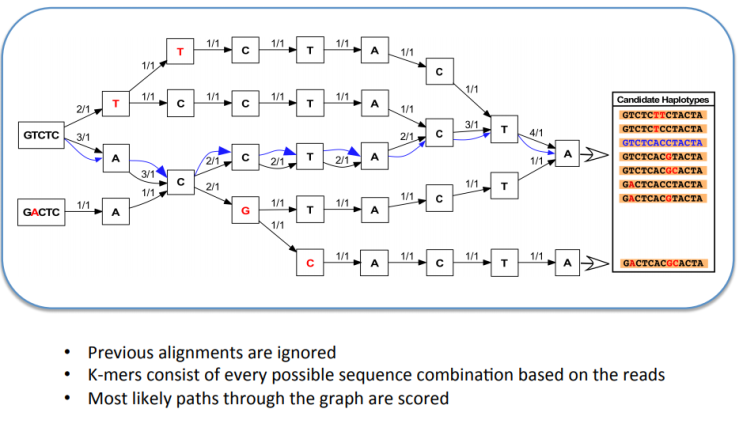
Seurat 에서 분석 진행 중 Normalized 단계가 있다. pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000) 이는 각 Cell 당 Feature(=gene)에 n수의 SD의 크기를 줄이기 위한 방법인데 그 중 default 값인 LogNormalize 에 대해 알아보고자 한다. normalization.method LogNormalize: Feature counts for each cell are divided by the total counts for that cell and multiplied by the scale.factor. This is then natural-log transformed using log1p.
이를 해석하면 이며 아래의 값을 통해 위 식이 맞는 것을 확인할 수 있다. > GetAssayData(pbmc, slot="counts")[c(74,...