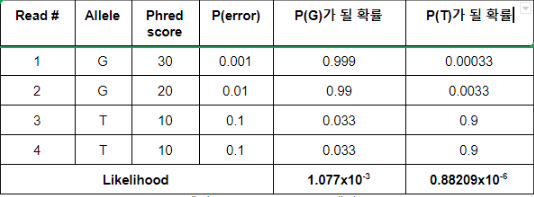
mutect2와 haplotype의 기본적인 원리는 같기에 seed(K-mer)가 더 많은 mutect2를 통해 raw_alignment.bam 과 re_alignment.bam 의 차이를 알아보고자 한다. reference에 alignment된 bam file의 크기가 12G 라고 가정해보자. realignment 된 bam file을 얻고싶어서 realignment tools을 사용하여 나오는 bam file의 크기는 600M 으로 약 1/20 이 줄어든다. * realignment tools 로 Mutect2 force calling mode 사용, (mutect2 force calling mode 추가옵션 : -L --force-active ture -active-probability-threshold 0.001 --max-reads-per-alignment-start 0 --min-base-quality-scrore 10 --pair-hmm-implementation FAST_AVAILABLE ) chr7에 한..........

원문 링크 : haplotypecaller (6) - result